La construction des tombes mégalithiques a cessé dans le nord-ouest de l'Europe à la fin du quatrième millénaire av. JC. pour des raisons inconnues. Ces tombes étaient nombreuses durant le Néolithique notamment dans le bassin parisien. C'était des tombes collectives avec différentes architectures selon la région de l’Europe. Dans le bassin parisien elles sont en forme de coffre allongé fait de dalles de pierre et elles sont appelées: allées sépulcrales. Le déclin à grande échelle de la construction de ces tombes à la fin du quatrième millénaire av. JC. pourrait être lié soit à un changement culturel soit à un déclin de la démographie. Les dernières recherches ont tendance à privilégier la seconde hypothèse. Les différentes raisons invoquées pour expliquer ce déclin démographique est soit une sur-exploitation de l'environnement aboutissant à une forte déforestation et à un appauvrissement des sols, soit des épidémies résultant de la proximité des hommes avec les animaux.
La tombe mégalithique de Bury est située à 50 km au nord de Paris. Il s'agit d'un exemple classique d'allée sépulcrale. Elle abrite les restes de 316 individus divisés en deux phases chronologiques successives. La première phase s'étend principalement entre 3200 et 3100 av. JC. Les corps y sont allongés sur le dos, la tête placée au sud et le corps aligné dans la direction de la tombe. La seconde phase est plus longue. Elle s'étend du début du troisième millénaire av. JC. jusqu'à 2470 av. JC. et se caractérise par des positions corporelles fléchies sur le côté sans orientation privilégiée:

Frederik Seersholm et ses collègues viennent de publier un papier intitulé: Population discontinuity in the Paris Basin linked to evidence of the Neolithic decline. Ils ont analysé le génome de 182 individus de l'allée sépulcrale de Bury appartenant aux deux phases d'occupation de cette tombe.
L'étude des sexes de ces individus montre qu'il y a beaucoup plus d'hommes que de femmes. Ainsi dans la phase 1, il y a 71% d'hommes et dans la phase 2, il y en a 73%. Ce déséquilibre concerne aussi bien les adultes que les enfants suggérant des traitements différentiés des sépultures selon le sexe. L'analyse des marqueurs uniparentaux montre une forte diversité mitochondriale durant les deux phases et une faible diversité des chromosomes Y. Alors que la phase 1 est caractérisée par les haplogroupes du chromosome Y: H2a1 et I2a1a2, la seconde est limitée à l'haplogroupe I2a2a2.
Les auteurs ont effectué une Analyse en Composantes Principales pour comparer ces différents génomes à d'autres anciens génomes de la région. Les résultats montrent que tous les individus de Bury se situent dans la diversité des fermiers d'Europe. Cependant ceux de la phase 1 (symboles marrons) sont très diversifiés alors que ceux de la phase 2 (symboles verts) sont plus regroupés entre eux:

Les auteurs ont ensuite analysé les segments Identiques par descente (IBD) pour étudier d'éventuels liens entre individus. Les résultats montrent que les individus se regroupent entre eux en fonction de leur phase suggérant deux populations indépendantes. La population de la phase 1 était de plus petite taille que celle de la phase 2. De plus les individus de la phase 1 montre des ascendances avec diverses populations du néolithique ancien d'Europe de l'ouest expliquant l'hétérogénéité de cette phase, alors que les individus de la phase 2 présentent une ascendance importante venue de la péninsule ibérique suggérant une migration vers le nord des fermiers du sud à partir de 2900 av. JC.
Les auteurs ont ensuite étudier les relations familiales entre individus. Les résultats montrent que les trois quarts des individus de la phase 1 sont reliés au premier ou au second degré alors qu'une part importante des individus de la phase 2 n'ont pas de relation familiale. De plus il n'y a aucun lien de parenté entre les individus de la phase 1 et ceux de la phase 2:

Les auteurs ont reconstruits 14 arbres généalogiques en tout, dont sept dans chacune des phases. Ces arbres varient en taille avec un grand qui s'étend sur 5 générations dans la phase 1 et des petits qui comprennent seulement deux individus. Dans la phase 1, aucun homme ne vient de l'extérieur: ils descendent tous d'autres individus de Bury. A l'opposé toutes les femmes sauf une de la phase 1 ont des liens familiaux qu'avec leurs enfants. Elles viennent toutes de l'extérieur suggérant une forte exogamie féminine dans cette société. L'arbre 1.A est le plus grand. Il est composé de 29 individus. Tous les hommes de cet arbre sont de l'haplogroupe du chromosome Y: H2a1. Trois frères forment la seconde génération. L'un deux a quatre fils dont deux ont leur génome séquencé. Les résultats de ces deux génomes montrent de longs segments homozygotes suggérant que leurs deux parents étaient reliés au troisième degré:

Dans la seconde phase il n'y a que deux femmes reliées au premier degré avec un autre individu: une mère et son fils et une femme avec son frère. Dans cette phase, une proportion plus élevée d'individus totalement sans lien de parenté suggère un type d'organisation différent avec un nombre beaucoup plus élevé de parents non biologiques inclus dans la tombe:

Tous ces résultats montrent que cette tombe a été utilisée par des société structurées différemment d'une phase à l'autre: de grandes familles dans la première phase et des plus petites dans la seconde avec un plus grand nombre d'individus sans aucune relation biologique. De plus il n'y a pas de corrélation entre les liens de parentés et la position des corps dans la tombe.
Les auteurs ont ensuite recherché d'éventuels pathogènes dans les génomes de ces individus. Ils ont ainsi mis en évidence quatre microbes: Yersinia enterolitica (n = 8), Yersinia pestis (n = 4), Borrelia recurrentis (n = 2) et Human alphaherpesvirus 1 (n = 1). Yersinia pestis est l'agent de la peste qui est présent chez trois individus de la phase 1 et un seul individu de la phase 2. Le pathogène Yersinia enterocolitica est responsable d'une entérite aiguë s'accompagnant de fièvre, diarrhées et douleurs abdominales. Le pathogène Borrelia recurrentis est responsable de fièvres récurrentes. Enfin Human alphaherpesvirus 1 est responsable de l'Herpès, une maladie contagieuse par simple contact buccal et responsable d'affection de la peau, des muqueuses et parfois du système nerveux, caractérisée par des crises d'éruption vésiculeuse de boutons groupés.
samedi 4 avril 2026
Discontinuité de la population du bassin parisien au Néolithique
samedi 4 avril 2026. ADN ancien
lundi 30 mars 2026
Le génome à haute couverture d'un Néandertal de l'Altaï
lundi 30 mars 2026. ADN ancien

Aujourd'hui, des données génétiques existent pour une trentaine de Néandertal. Seulement trois d'entre eux sont des génomes à haute couverture: une femme de la grotte Denisova dans l'Altaï vieille de 120.000 ans (Denisova5), une femme de la grotte Chagyrskaya également dans l'Altaï vielle de 80.000 […]
samedi 7 février 2026
Relations entre les communautés de chasseurs-cueilleurs et de fermiers sur l'île de Gotland en Suède
samedi 7 février 2026. ADN ancien

La Scandinavie a été peuplée initialement par des groupes du Mésolithique venus de l'ouest (WHG) et du nord-est (EHG) qui se sont mélangés génétiquement résultant en différents groupes de chasseurs-cueilleurs Scandinaves hébergeant des proportions WHG et EHG diverses. La néolithisation de cette […]
samedi 17 janvier 2026
La sépulture campaniforme de Kolbsheim Knoblochsberg
samedi 17 janvier 2026. Archéologie

En 2017, une nouvelle sépulture campaniforme a été découverte sur le site de Knoblochsberg localisé dans la commune de Kolbsheim à l'ouest de Strasbourg suite à un diagnostic réalisé par Archéologie Alsace. Cette sépulture a été réétudiée par la suite par Antea Archéologie. Les informations […]
vendredi 26 décembre 2025
Génomes anciens dans les Pouilles en Italie du sud durant l'âge du bronze
vendredi 26 décembre 2025. ADN ancien

Roca Vecchia, localisé dans les Pouilles sur la côte Adriatique, est l'un des plus importants sites fortifiés de l'âge du bronze moyen de la région. Ce site a commercé avec les cultures de la mer Égée. Ainsi la découverte de céramiques Égéennes suggère que des potiers Crétois ou de Grèce […]
samedi 20 décembre 2025
Anciens génomes dans une communauté de l'Âge du Bronze d'Italie du sud
samedi 20 décembre 2025. ADN ancien

La Calabre est la région la plus méridionale d'Italie continentale, bordée au sud et à l'est par la mer Ionienne et à l'ouest par la mer Tyrrhénienne. La région a une histoire complexe de migrations anciennes et récentes. Au néolithique, une population d'Anatolie a migré par bateaux en suivant la […]
dimanche 14 décembre 2025
Le cimetière campaniforme de Reguisheimer Feld à Ensisheim en Alsace
dimanche 14 décembre 2025. Archéologie

En 2017 et 2018 un cimetière campaniforme a été découvert sur le site de Reguisheimer Feld à Ensisheim en Alsace, lors d'une fouille préventive au préalable à l'établissement d'une zone d'activité, et réalisée par Archéologie Alsace. Les informations suivantes sont issues du rapport de fouille […]
dimanche 26 octobre 2025
Génome d'un dénisovien de l'Altaï vieux de 200.000 ans
dimanche 26 octobre 2025. ADN ancien

Les dénisoviens sont un groupe humain éteint qui a été identifié pour la première fois en 2010 à partir du génome d'une petite phalange d'un doigt découverte dans la grotte Denisova dans les montagnes de l'Altaï situées à l'ouest de la Sibérie. Cette première analyse a montré que les dénisoviens […]
samedi 18 octobre 2025
Anciens génomes de l'est du Kazakhstan entre le début du Néolithique et la fin de l'Âge du Bronze
samedi 18 octobre 2025. ADN ancien

Les populations de l'Asie Intérieure sont remarquablement diverses bien que principalement structurées le long de trois gradients reflétant différentes zones écologiques et géographiques. Les récentes études de paléo-génomique ont permis de reconstruire les différents processus démographiques de la […]
jeudi 2 octobre 2025
Mélange génétique entre les populations associées au Gravettien d'Europe de l'Est et de l'Ouest en Europe
jeudi 2 octobre 2025. ADN ancien
Le Gravettien est une ancienne culture paléolithique datant d'avant le dernier maximum glaciaire. Malgré une certaine similarité de la technologie, la culture Gravettienne n'était pas totalement homogène. Ainsi neuf groupes régionaux ont été identifiés archéologiquement. Les génomes Gravettiens […]
jeudi 25 septembre 2025
Une nouvelle découverte d'une sépulture campaniforme individuelle à Saint Martin la Garenne
jeudi 25 septembre 2025. Archéologie

Le musée de la préhistoire d'Île de France à Nemours présente actuellement une exposition intitulée: Pouvoir et métal - L'âge du Bronze en Île-de-France, de 2300 à 800 avant notre ère. Cette exposition démarre sur l'utilisation de l'or et du cuivre à la fin du Néolithique. A cette occasion elle […]
samedi 30 août 2025
Génomes provenant d'une communauté de la fin de l'âge du bronze enterrée dans un tumulus du nord-est de la péninsule Ibérique
samedi 30 août 2025. ADN ancien

La diversité génétique européenne a été façonnée par différents événements démographiques clés, dont la dernière transformation majeure était l'afflux de migrants issus de pasteurs de la steppe ponto-caspienne entre 3000 et 2000 av. JC. A l'âge du bronze, divers groupes culturels ont émergé et se […]
lundi 11 août 2025
Étude paléogénétique d'une nécropole de la fin de l’Antiquité et du début du Moyen Âge à Angers
lundi 11 août 2025. ADN ancien

Traversée par la Maine peu avant sa confluence avec la Loire, la ville d'Angers est occupée par l'homme depuis au moins le néolithique. À la fin de l’Antiquité, la cité des Andécaves, ancien peuple gaulois, est l’un des derniers territoires qui perdure sous l’Empire romain et reste aujourd’hui une […]
mercredi 23 juillet 2025
Histoire génomique de l'Italie du nord-est entre le mésolithique et l'âge du bronze
mercredi 23 juillet 2025. ADN ancien

Les Alpes italiennes de l'est ont joué un rôle de pont entre les populations du nord des Alpes et celles du sud des Alpes. Durant le dernier maximum glaciaire les Alpes étaient recouvertes de glaciers. Après 18.000 ans les glaciers se sont retirés peu à peu laissant la place à une population de […]
vendredi 4 juillet 2025
Génome d'un individu de l'Ancien Empire Égyptien
vendredi 4 juillet 2025. ADN ancien

La civilisation égyptienne a développé une architecture monumentale et un système de croyances stable pendant des millénaires entre 3150 et 30 av. JC. A la suite de la réunification politique de l’Égypte à la fin du quatrième millénaire av. JC., l'Ancien Empire a connu des progrès considérables dont […]
mardi 24 juin 2025
L'homme de Harbin en Chine est un Dénisovien
mardi 24 juin 2025. Protéome ancien
Les Dénisoviens sont un groupe d'anciens hominidés d'Asie identifié à partir de l'analyse ADN de fossiles issus de la grotte Denisova située en Sibérie. Les fossiles actuels attribués à l'homme de Denisova représentent quelques dents et quelques fragments d'os. Des analyses protéomiques précédentes […]
samedi 21 juin 2025
ADN ancien en Colombie entre 6000 et 500 ans
samedi 21 juin 2025. ADN ancien
Les études précédentes de paléo-génétique ont montré que les populations Amérindiennes en dehors de la zone arctique, dérivent d'un mélange génétique entre une population est asiatique et une population sibérienne qui a eu lieu quelque part dans le nord est de l'Asie il y a plus de 20.000 ans. Il y […]
dimanche 15 juin 2025
Transition entre le Néolithique et l'Âge du Bronze à Mas d’en Boixos en Catalogne
dimanche 15 juin 2025. ADN ancien
Mas d’en Boixos est le site archéologique le plus significatif de la région de Penedès en Catalogne située entre Barcelone et Tarragone. Les fouilles débutées en 1997 ont livré 450 structures datées entre le début du Néolithique et la fin de l'Âge du Fer soit entre 5500 et 200 av. JC. Ce site est […]
samedi 3 mai 2025
Continuité génétique dans la culture des Pueblos au Nouveau Mexique
samedi 3 mai 2025. ADN ancien

Les anciens pueblos créés par la culture ancestrale des Pueblos, un terme indiquant leur relation ancestrale avec les tribus amérindiennes contemporaines des Pueblos, comptent parmi les cultures archéologiques les plus reconnaissables d'Amérique du Nord. De nombreux pueblos anciens sont devenus le […]
vendredi 25 avril 2025
Anciens génomes des communautés Phéniciennes et Puniques
vendredi 25 avril 2025. ADN ancien
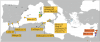
La culture phénicienne a émergé dans des cités états de l'Âge du Bronze du Levant. Au début du premier millénaire av. JC., les phéniciens ont établi un vaste réseau commercial le long des côtes méditerranéennes jusque sur les côtes sud-ouest de la péninsule Ibérique, diffusant leur culture, leur […]
« billets précédents - page 1 de 34
